Kore Huntington
Daha geniş anlamda eş anlamlılar
- Vitus dansı (vulg.)
- Huntington hastalığı
İngilizce: Huntington hastalığı, büyük kore.
Tanım
Kalıtsal hastalıkyıkıma götürmek Beyin hücreleri bilinçsiz tutma ve destekleme motor becerilerinin belirli beyin bölgelerinde. Hastalık genellikle 35-50 yaşları arasında ortaya çıkar. Yıl ve ifade edilir
- Uzuvların kasıtsız, yıldırım hızında, savrulma hareketleri gibi hareket bozuklukları
- grimacing
- Entelektüel kapasitenin bozulması ve
- Kişilikte düşüş.
Huntington hastalığının yaşam beklentisi nedir?
Normal popülasyonla karşılaştırıldığında, Huntington hastalığı olan hastalarda yaşam beklentisi önemli ölçüde azalır. Genel yaşam beklentisinin ne kadar yüksek olduğu kişiden kişiye büyük ölçüde değişir. Bu, bir yandan başlangıç yaşına ve diğer yandan hastalığın seyrine bağlıdır. Genellikle ilk belirtiler 30 ile 40 yaşları arasında ortaya çıkar. Etkilenenlerin neredeyse yarısı hastalığın ilk 10 yılında hayatını kaybediyor. Hastalığın 15. yılından sonra sadece% 25'i hala yaşıyor. Bununla birlikte, vakaların% 10'unda hastalık da 20 yıldan fazla sürdü. Temel olarak, kadınların ortalama olarak erkeklerden biraz daha uzun bir hastalık süresi vardır. Hastalık ne kadar erken ortaya çıkarsa, seyir o kadar şiddetli olur. Huntington hastalığı olan hastaların yaşam beklentisi ortalama olarak 40 ila 50 yıl arasındadır, ancak hastalığın geç başlangıcıyla yaklaşık 60 yaşına ulaşılabilir.
Epidemiyoloji:
Huntington hastalığının sıklığı 5 - 10 / 100.000 olarak verilir, kalıtım otozomal baskındır. Bu, etkilenenlerin çocuklarının hastalığı kendilerinin geliştirme riskinin% 50 olduğu anlamına gelir.
Semptomlar:
Etkilenenler kas gevşekliğini yaşarlar ve aynı zamanda genişler. yıldırım gibi savrulma hareketleri Duygusal gerginlik olduğunda ağırlaşan ve nadiren uyku sırasında ortaya çıkan uzuvların.
Bunun nedeni, gerekli dürtülerin hareketi engelleyememesidir. Hareketlerin bozulmuş koordinasyonu, yüz buruşturma, yutma bozuklukları ve konuşma güçlüklerinde daha da ifade edilir. Kore Huntington ilerledikçe, hasta ilerledikçe yürümekte güçlük çeker, göz hareketlerini koordine eder ve dışkı ve idrarı tutamaz hale gelir.
Kore ile de ortaya çıkar Kişilik değişiklikleri öfke nöbetleri ve dikkat bozuklukları gibi psikozlar bağlamında aldatmalar gibi. Entelektüel performanstaki düşüş, ilerici bunaklık (Kazanılmış zihinsel engel, oraya bakın). Huntington hastalığı, genellikle hastanın genel durumunun kötü olmasının neden olduğu ikincil hastalıkların bir sonucu olarak, teşhisten sonraki 15-20 yıl içinde ölümcüldür.
İlk işaretler nelerdir?
Huntington hastalığının ilk belirtileri genellikle 30 ila 40 yaşları arasında fark edilir. Psikolojik şikayetler çoğu zaman hastalığın özelliği olan hareket bozukluklarından yıllarca önce gelir. Tipik psikolojik anormallikler depresyon ve azalmış dürtüdür. Bazen başlangıçtaki bilişsel eksiklikler konsantrasyon ve hafıza bozuklukları şeklinde kendini gösterir. Bu belirtiler erken dönemlerde kolaylıkla depresyonla karıştırılabilir. Hastalığın sıklıkla diğer insanlara karşı dürtüsel ve incitici davranışlara yol açması, akrabalar için de streslidir.
Hastalar kısmen görsel bilgiler alabilir, örn. Yüz ifadeleri artık doğru şekilde işlenmez ve bu nedenle başkalarının duygularına artık uygun şekilde tepki vermez. Hareket bozuklukları başlangıçta şu şekilde karakterize edilir: hiperkinezi (Yunanca hiper - hakkında, kinesis - hareket). Bu, istenmeyen hareketlerin artması anlamına gelir. Kas tonusu - kaslardaki gerginlik durumu - azalır. Hastalar kendi bedenleri üzerindeki bu kontrol eksikliğini çok stresli bulurlar. Bazen, özellikle erken dönemde İntihar girişimleri.
Hastalık nasıl ilerliyor?
Huntington hastalığı bir kronik ilerleyen nörodejeneratif Hastalık. Bu, genellikle yavaş ama sürekli ilerlediği, sinirleri tahrip ettiği ve nihayetinde hastanın ölümüne yol açtığı anlamına gelir. Psikolojik anormalliklere ek olarak, hareket bozuklukları da hastalığın karakteristiğidir. Erken dönemlerde genellikle daha fazla istenmeyen hareketler (hiperkinezi) üzerinde. Zamanla biri gelişir hipokinezi. Kelimenin tam anlamıyla tercüme edildiğinde, bu "daha az egzersiz" anlamına gelir, kastedilen, Parkinson sendromunda da tipik olduğu gibi egzersiz eksikliğidir. Hastalık ilerledikçe, hasta giderek daha fazla bakıma muhtaç hale gelmektedir. İlerleyen demans başlangıçta dilin zayıflamasına ve yönelim bozukluğuna yol açar. Yiyecek alımı genellikle yutma bozuklukları nedeniyle zorlaşır ve hastalar kilo verir. Ortalama olarak hastalar ölür 10-15 yıl hastalığın başlangıcından sonra. Hastalığın başlangıcı geç ortaya çıkarsa, hastalığın seyri genellikle biraz gecikir.
Tedavi var mı?
Şu anda Huntington hastalığının tedavisi yoktur. 1993'ten beri hastalığın nedeninin kusurlu bir gen olduğunu biliyoruz Kromozom 4. Ne yazık ki, şu anda genetik kusuru veya sonuçlarını herhangi bir şekilde tedavi etmenin bir yolu yok. Dolayısıyla bu noktada hastalığın seyrini durduramazsınız. Elbette, yeni terapötik yaklaşımlar konusunda yoğun araştırmalar var. Hastalığın genetik temeli artık iyi bilinmektedir.Bu nedenle, etkilenenler ve yakınları, yalnızca araştırmanın bir noktada önemli bir atılım yapacağını umabilirler.
Hangi ilaçlar yardımcı olur?
Huntington hastalığına bir gen mutasyonu neden olur. Ne yazık ki şu anda bu nedeni tedavi eden veya hastalığı iyileştiren hiçbir ilaç bulunmamaktadır. Farklı semptomları ilaçla tedavi etmeye çalışabilirsiniz. Nöroleptikler genellikle klasik hareket bozukluklarına karşı kullanılır. Antidepresanlar depresif ruh hallerine yardımcı olur. Sonuçta, bu ilaçlar hastalığın ilerlemesini durduramaz. İlaçla semptomları biraz daha iyi kontrol etmeye çalışıyorsun.
Son aşama neye benziyor?
Genellikle son aşamadır 10-15 yıl hastalık başladıktan sonra ulaşıldı. Hastalar yatalaktır ve günün her saati bakıma ihtiyaçları vardır. Hastalık ilerledikçe gelişen yutma bozukluğu nedeniyle, çoğu çok zayıflamıştır (tıbbi: kaşektik). Ayrıca yiyecek yutulursa, yaşamı tehdit eden zatürre için kalıcı bir risk vardır (Aspirasyon pnömonisi) geliyor. Hasta artık yutamıyorsa yapay beslenme düşünülmelidir. Hastalık ilerledikçe psikolojik anormallikler de artar. Sonunda demans ilerledi, hastalar iletişim yeteneğini kaybediyor ve yönünü şaşırıyor.
Ayırıcı tanılar
Hareket bozuklukları ve zihinsel gerilemeden oluşan benzer semptomlar, Creutzfeld-Jakob hastalığıhastalığın sonraki aşamalarında Frengi ve iltihaplanmadan sonra Beyin meydana gelir.
Huntington hastalığına ne sebep olur?
Huntington hastalığı genetik bir hastalıktır. Nedeni genetik bir kusurdur. Protein (protein) hastalığa neden olan hunttin denir. Bunu kodlayan gen, Kromozom 4. Huntingtin proteininin mutasyonu, belirli beyin bölgelerinde özel sinir hücrelerinin ölmesine neden olur. Bu, yavaş ilerleyen bir süreçtir, bu nedenle hastalık sözde biridir. nörodejeneratif hastalıklar. Hastalıkla bağlantılı birçok patolojik süreç henüz tam olarak araştırılmamıştır. Bununla birlikte, Huntington hastalığının biri olduğu bilinmektedir Trinükleotid hastalığı davranır. Sağlıklı insanlarda DNA'da 20 kata kadar belirli bir üçlü kombinasyon tekrarlanır. Huntington hastalığı olan hastalarda bu kombinasyon 60 ila 250 kez olmak üzere çok daha sık tekrarlanır. Sonuç olarak, gen artık doğru bir şekilde okunamaz ve Huntingtin proteini yanlış bir şekilde birleştirilir. Bu tekrar ne kadar çok olursa, kişi semptomları o kadar erken yaşar. Bir hastada ne kadar çok tekrar tespit edilebilirse, hastalık o kadar zorlaşır.
Teşhis:
Ailede Huntington hastalığının ortaya çıkışıyla ilgili tıbbi geçmiş ve soruların toplanması. Sinir sistemine odaklanan fizik muayene.
Beyin aktivitesi ölçümü (EEG), muhtemelen kafanın bilgisayarlı tomografisi (dilim röntgeni). Genetik materyaldeki temel değişiklikler bilindiğinden, genetik bir test, Huntington hastalığını güvenilir bir şekilde teşhis edebilir ve hatta tahmin edebilir. Bununla birlikte, bu tür bir öngörücü (öngörücü) teşhis, hastalık şu anda tedavi edilemediğinden ve bu nedenle hiçbir terapötik sonuç olmayacağından son derece nadiren faydalıdır.
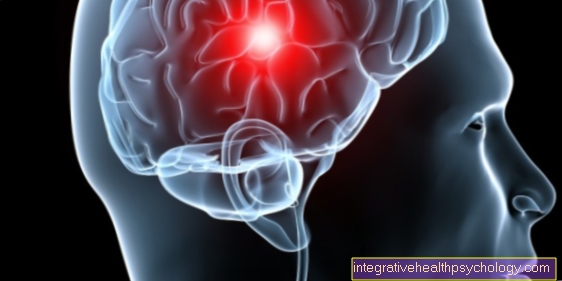
Beynin MR görüntüsü
Huntington hastalığından şüpheleniliyorsa, beynin kesitsel bir görüntüsünün alınması mantıklıdır. Hastalık bir nörodejeneratif İşlem sırasında belirli beyin bölgelerindeki sinir hücrelerinin öldüğü hastalık. Bu aynı zamanda MRI görüntülerinde de görülebilir. Doku atrofisi, özellikle gönüllü hareketten sorumlu olan bölgede belirgindir. Bu nasıl Lateral ventrikül (= serebral su ile dolu boşluklar) görüntülemede genişledi. Bu, Huntington hastalığı için nispeten klasik bir bulgudur. Tanısal kesinlik, genetik test ile sağlanır (bununla ilgili bölüme bakın).
Huntington hastalığı nasıl kalıtsaldır?
Huntington hastalığı bir otozomal dominant kalıtsal hastalık. Bir gen baskın olarak kalıtsalsa, zaten kusurlu olduğu anlamına gelir. allel ikisinden birinde kromozomlar karakteristik ifadeye yol açar. Otozomal terimi, otozomlardan türetilmiştir. Cinsiyet belirlemede yer almayan tüm kromozomlara otozom denir. Bu, mirasın cinsiyetten bağımsız olduğu anlamına gelir. Böylece, kusurlu geni her iki ebeveynden de miras alabilirsiniz. Bu nedenle erkekler ve kadınlar eşit derecede etkilenir. Huntington hastalığı durumunda, kusurlu gen açık Kromozom 4. Kalıtım cinsiyetten bağımsız olmakla birlikte, kusurlu gen babadan miras alınırsa hastalığın daha erken başladığı ve daha dramatik bir seyir izlediği gösterilmiştir. Anne kalıtımı durumunda ise, hastalığın başlangıcının daha sonra ortaya çıkması daha olasıdır.
Genetik test
Huntington hastalığından sorumlu olan mutasyona uğramış gen açığa çıktı Kromozom 4. 1993 yılında keşfedildi. O zamandan beri bir genetik test mevcuttur. Dolayısıyla, bir hastanın Huntington hastalığına sahip olduğundan şüpheleniliyorsa, hastanın DNA'sında bu mutasyona sahip olup olmadığını görmek için bir kan örneği incelenebilir. Bu teşhisi güvence altına alır. Huntington hastalığı olan sevdiklerine sahip sağlıklı insanlar da mutasyon için kanlarını test ettirebilirler. Huntington hastalığı kalıtsal bir hastalıktır. Bunun genellikle etkilenenlerin yaşamı için geniş kapsamlı sonuçları vardır, bu nedenle sağlıklı insanlarda genetik testler için özel yönergeler vardır. Örneğin. reşit olmayanlar test edilmemiştir; üçüncü şahısların (ebeveynler, partnerler, ...) talebi üzerine hiçbir genetik test yapılamaz. Sağlıklı insanlarda gen mutasyonunu tespit ederek, kişi hemen teşhis edilmez, ancak DNA'daki belirli bir dizinin belirli sayıda tekrarına ulaşılırsa, etkilenen kişi büyük olasılıkla hastalık sırasında Huntington hastalığını geliştirecektir.
Terapi:
Huntington hastalığının nedeninin tedavisi şu anda mümkün değildir. Aşırı hareket bozuklukları ilaçla bastırılabilir. Belirli koşullar altında, psikoterapiye eşlik etmek veya bir kendi kendine yardım grubuna katılmak, hastanın hastalık hakkındaki bilgilerini işlemesine yardımcı olabilir.
bunaklık
Huntington hastalığı, klasik hareket bozukluklarının yanı sıra psikolojik değişikliklere de yol açar. Bunlar, Etkilemek (= Ruh hali depresyona kadar yükselir), ama aynı zamanda bilişsel sınırlamalar. Bunlar genellikle erken evrelerde hafıza bozuklukları olarak ortaya çıkar. Hastanın entelektüel yetenekleri başlangıçta çok az bozulmuştur; bu genellikle yabancılar tarafından fark edilmez. Hastalık ilerledikçe, bunamaya kadar bilişsel becerilerde artan bir kayıp olur. Konuşma zayıflaması meydana gelir ve hastalar genellikle tamamen kafasını karıştırır.